细胞注释小tips
关于细胞注释
首先应当分清楚是否需要对合并的数据进行批次处理(往往是需要进行批次处理的)
就拿我这次处理的数据作一个举例。
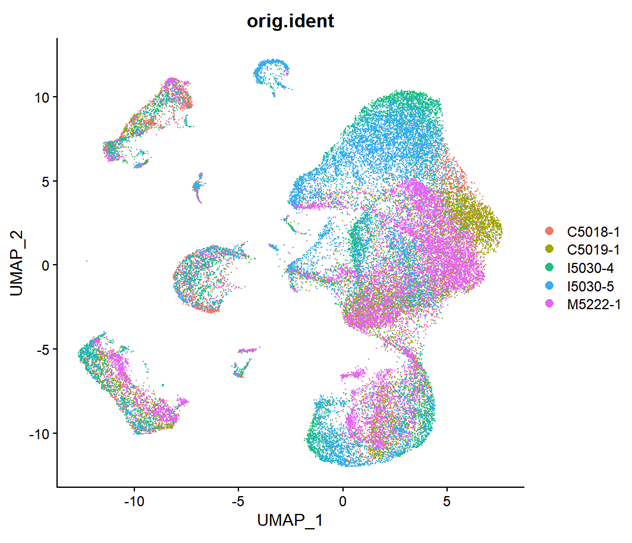
可以看出在上图中不同批次的细胞的颜色分布是不太均匀的。尤其是上面的一簇,基本是蓝色的,中间偏右的一大簇里面也可以看到有较为明显的界限。接下来看看我使用seurat包处理过后的数据图像。
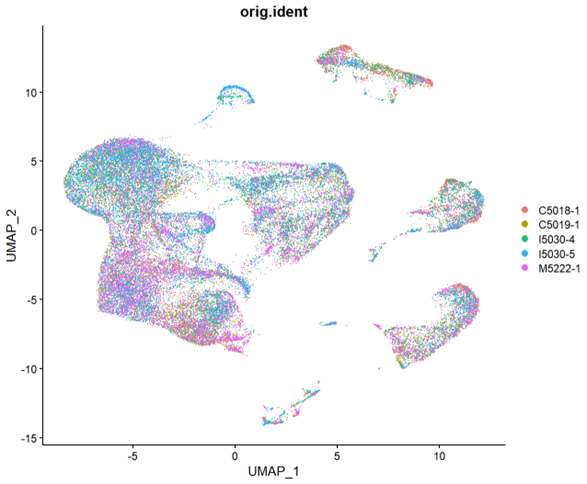
可以看到,上图的图像中各个批次的分布明显比第一张图像更为均匀。
根据论文以及专业方向老师给的genemarker手动标注后,可采用Enrichr网站进行进一步的校对(可将一个cluster的所有genemarker都复制进去进行校对)。
猛然回首,自己进入单细胞分析的领域已经快半年了。单是整合的一步骤就有许多的小操作,我们需要控制整合过程中的features,控制anchor的大小。总之整个过程也是一个需要调参的过程。目前还有一项技术能够评估整合停止的参数究竟在什么时候是最好的。
对于一些无论如何调整resolution(分辨率)都无法达到理想的分群的情况,我们可以采取讲某一群单独取出来以后进行细胞注释,得到对应细胞的idents以后再在原来的数据中进行赋值。
以下为一个例子:
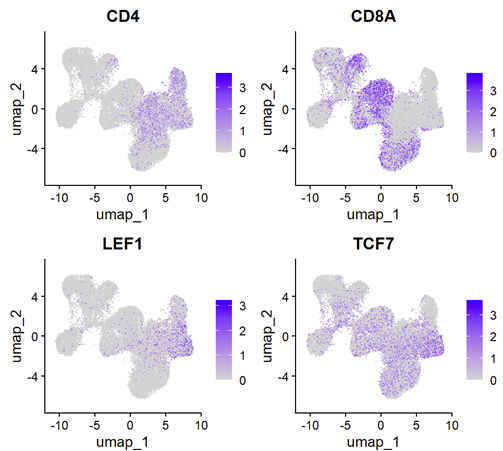
通过featureplot可以看到图中的右下角的一小块应该是cd8_naive,但是上图的分群(resolution=5)也没有找到一个合适的分群。故采用以上所说的办法。
先贴代码:
subcd8naive<-subset(subseu, subset=celltype=='CD8A_naive_TCF7')
DimPlot(subcd8naive, reduction = 'umap')
subcd8naive<-RunUMAP(subcd8naive, dims = 1:35)
DefaultAssay(subcd8naive) <-'integrated'
subcd8naive<- FindNeighbors(subcd8naive, dims = 1:35)
subcd8naive <- FindClusters(subcd8naive, resolution = 1.8)
DimPlot(subcd8naive, label = T)
DefaultAssay(subcd8naive) <- "RNA"
subcd8naive <- JoinLayers(subcd8naive)
markers <- FindAllMarkers(subcd8naive, only.pos = TRUE, min.pct = 0.25,
logfc.threshold = 0.25)
write.xlsx(markers, file='cd8marker.xlsx')
FeaturePlot(subcd8naive,features = c('CD4','CD8A','LEF1','TCF7'), order = F)
new.cluster.ids <- c(new.cluster.ids <- c('CD8A_naive_TCF7','CD4_Tfh_CD40LG','CD4_Tfh_CD40LG','CD4_Tfh_CD40LG',
'CD4_Tfh_CD40LG','CD4_Tfh_CD40LG','CD4_Tfh_CD40LG','CD4_Tfh_CD40LG'))
names(new.cluster.ids) <- levels(subcd8naive)
subcd8naive <- RenameIdents(subcd8naive, new.cluster.ids) #修改Idents
#在metadata中,添加Celltype信息
subcd8naive$celltype <- Idents(subcd8naive)
cd4<-WhichCells(subcd8naive, idents='CD4_Tfh_CD40LG')
subseu@meta.data[cd4,]$celltype <- "CD4_Tfh_CD40LG"当然,上述代码只是我针对这个数据集所做的操作,一些具体的数值还需要根据不同的数据集作调整。基本的步骤是:取出应该调整的部分所在的群,重新跑UMAP图,然后对数据进行重新分群,再根据新的featureplot进行注释,找出目标(cd8_naive)的位置以及idents(cd4),再将其重新复制到大的数据中去。
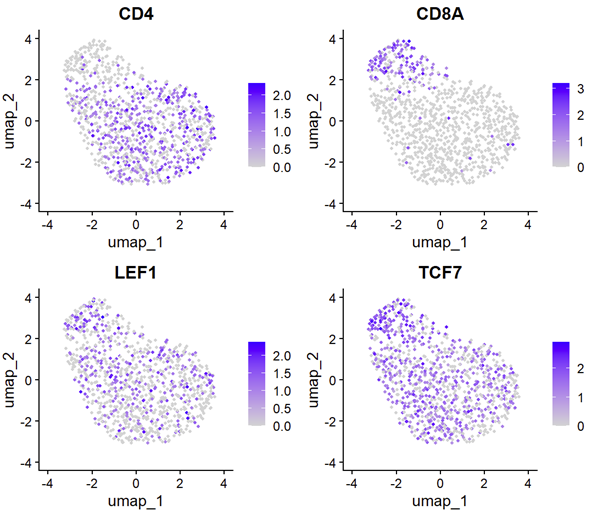
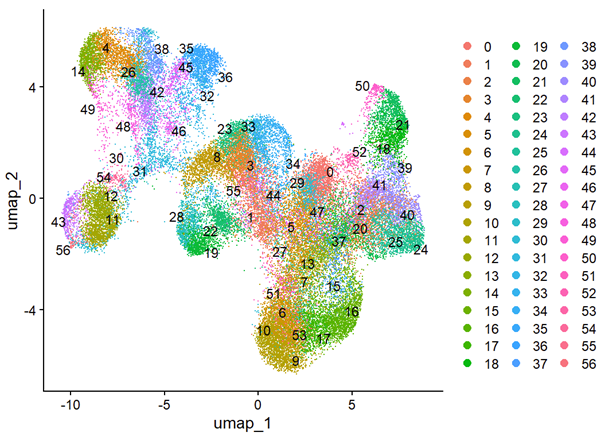
上图为取出后重新绘制featurePlot的中间结果图。
最后再贴上最终的处理结果(具体是针对原始分群中的第20群进行处理):

如果对贴出的代码有任何问题可以联系我的邮箱:math_jenris@163.com